
The Center for Cell Fate and Therapeutics
The Center for Cell Fate and Therapeutics is a multidisciplinary, interdepartmental program focused on understanding the mechanisms that maintain cell identity, fate and plasticity and how derangement of these processes lead to disease. This knowledge will be applied to preserve human health.
Using cutting-edge basic, clinical and translational science the Center will pursue the following general goals:
- Generate fundamental new knowledge on cell fate and plasticity.
- Accelerate the transfer of knowledge to excel in clinical practice and maintain the health of individuals and their communities.
- Collaborate with the private sector, government and industry to develop new therapies and tools.
- Educate the new cadre of graduate students and physician-scientists.
- Engage our communities and leaders in implementing novel educational -health practices throughout the State and globally.
- Develop international partnerships to enhance our global reach.
Areas of endeavor
The Center for Cell Fate and Therapeutics is devoted to understand how cells acquire and maintain their identity, how cells organize to form healthy tissues and maintain homeostasis, and how maladaptive changes in cell fate result in disease.
The center will also undertake a systems biology-public health approach to understand how environmental, cultural and socio-economic patterns result in epigenetic changes driving health disparities and individual responses.
Furthermore, the center will use the knowledge gained at the bench and clinics to develop diagnostic and prognostic markers, therapeutic strategies and preventive measures to benefit individual patients and their community.
As shown in Figure 1, efforts directed at understanding and manipulating how cells acquire and maintain their identity will be crucial to prevent and cure disease. A few examples of the disorders that can benefit from this knowledge are shown below. Ultimately, knowledge gained from the studies described herein will lead to novel biomarkers and individualized, targeted, molecular and cell therapies to maintain the health of our communities. Experimental approaches will range from examination of epigenetic and molecular mechanisms that govern cell lineage and fate to organismal function and population studies during development and disease. Therefore, the scientific inquiry is not limited to molecules and cells and exploits such knowledge to improve the lives of individuals and their community at a local and global scale.
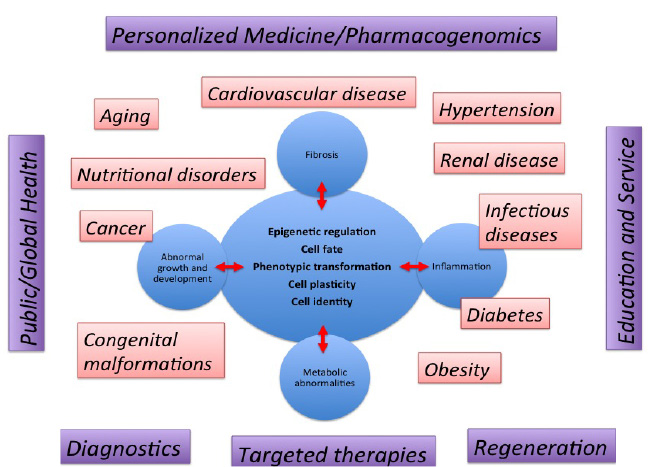
Research Themes
Early development of the kidney Vasculature.
Maria Luisa S. Sequeira Lopez, MD
Minghong Li, MD: Foxd1 gene and patterning of the kidney vasculature
Yan Hu, Graduate Student: Fate of hemangioblast in heart and kidney.
Ariel Gomez: Clonal analysis of vascular formation
As tissues and organs form, they require oxygen and nutrients, which are transported to the growing cells by blood vessels. Thus, the vasculature is crucial for the successful development and morphogenesis of the embryo and later in life to maintain the vitality of all our organs.
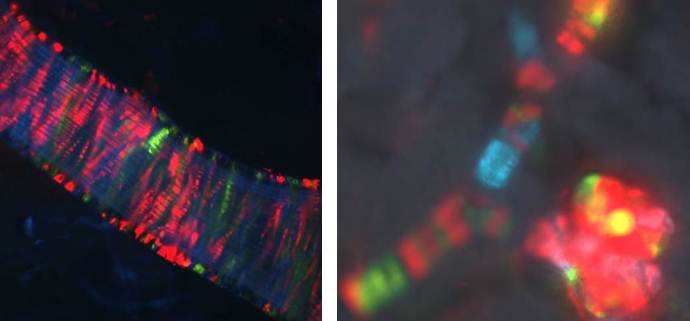
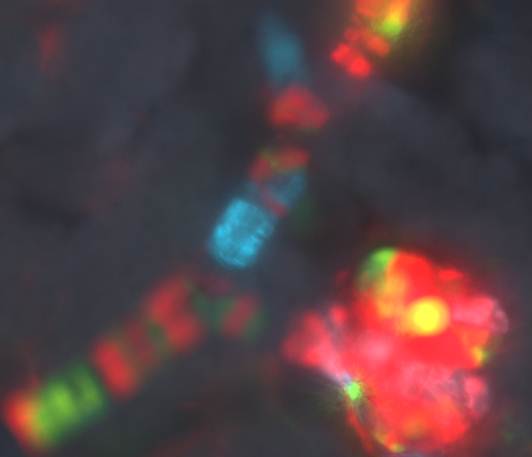
Work in the Sequeira-Lopez lab demonstrated that blood vessels in the embryo form by a process of hemovasculogenesis, the simultaneous formation of vessels and their own blood cells. Using in vivo lineage tracing techniques (cre-lox) her group have led to the successful identification of the mesenchymal precursor cells that form all the kidney vasculature. More recently, using a “confetti” mice crossed to Foxd1-cre –expressing mice, she resolved and old-standing scientific question by demonstrating that the cells that compose the kidney vasculature are composed of an overall mosaic, non-clonal population, interspersed with “clonality bands”, fitting perfectly a model of vasculogenesis (Figure 2). This fundamental discovery has application for the understanding of developmental processes and tissue regeneration because it indicates that the kidney has intrinsic precursors that can be coaxed to develop in situ. Cancerous cells also utilize many of the processes involved in vessel development when they metastasize outside their place of origin. In essence cancerous cells highjack the host vasculature to promote their own uncontrolled growth.
{kind=link}
Figure 2: Clonal banding and mosaicism in the kidney vasculature: To the left a major intrarenal artery. Individual cells (stripes) are genetically labeled randomly with different colors. Cells bearing the same color and in proximity to each other arise from the same clone. To the right a smaller afferent arteriole and its glomerulus. Arterioles are composed of a mosaic of cells from separate clones. Although there are bands of clonality, the overall architecture of the vessel is a mixed population agreeing with our model of vessel formation by a process of vasculogenesis. The glomerulus shown however is populated mostly with cells from the same clone.
ugene Lin, Graduate Student
Maria Luisa Sequeira Lopez, MD
Ariel Gomez, MD
The mechanisms underlying the establishment, development, and maintenance of the renal vasculature are poorly understood. Here we propose that the transcription factor recombination signal binding protein for immunoglobulin kappa J region (RBP-J) plays a key role in the differentiation of the mural cells of the kidney arteries, as well as the mesangial cells of the glomerulus. Deletion of RBP-J in cells of the forkhead box D1 (Foxd1) lineage, which differentiate into the entirety of the kidney arterial tree along with mesangial cells and pericytes, resulted in significant kidney abnormalities and mortality by day 30 post-partum. In newborn mutant animals we observed a decrease in the total number of arteries and arterioles, along with thinner vessel walls. This was accompanied by depletion of renin cells. Nephrons displayed striking abnormalities, including a failure of Foxd1 cells to populate the glomeruli, and absence of mesangial cells (Figure 3), and in extreme cases a complete loss of interior structure and the development of glomerular aneurysms. By around time of death the kidney malformations were accentuated by extensive fibrosis and glomerulosclerosis. In summary, we conclude that RBP-J is essential for proper formation of the kidney vasculature and glomeruli.
Control Foxd1-cre; RBP-J
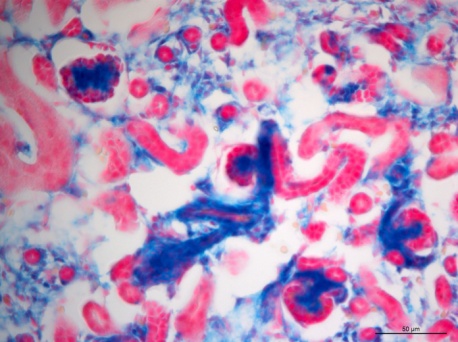
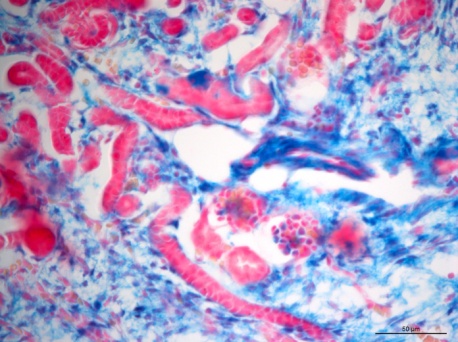
Figure 3: Lineage study of Foxd1 cells upon RBP-J deletion. In Control kidneys from newborn mice, Foxd1-lineage cells (labeled blue by the Xgal lineage tracer)) can be found in undifferentiated stroma, interstitium, the mural cell layer of arterioles and mesangial cells (blue). Foxd1RBPJ-/- animals lose expression of the reporter within glomerular mesangial cells.
Silvia Medrano, Ph.D.
Ariel Gomez, MD
Congenital kidney diseases are the most common causes of renal failure in infants. Of these, obstructive nephropathy is considered the most prevalent.
Many cases of obstructive nephropathy do not have a demonstrable anatomical obstruction appearing in early fetal life. The term “functional” hydronephrosis has been coined to denominate this type of obstruction. Although functional hydronephrosis can have equally devastating consequences as physical obstruction leading to renal failure, the underlying causes of functional hydronephrosis are not well understood. We discovered that microRNAs may hold the secret to this poorly understood disease. MicroRNAs are small, non-coding RNAs that regulate the expression of target genes at the post-transcriptional level. We found that miR-145, a member of the miR-143/145 cluster highly expressed in smooth muscle cells of renal arterioles, was also present in the pelvicalyceal system and the ureter.
To evaluate whether the miR-143/145 cluster is involved in urinary tract function we performed morphological, functional, and gene expression studies in mice carrying a whole body deletion of miR-143/145. miR-143/145-deficient mice did not exhibit overt morphological changes in the kidney vasculature. However, these mice developed hydronephrosis, characterized by severe papillary atrophy and dilatation of the pelvicalyceal system without obvious anatomical obstruction. To determine whether ureteral peristalsis was affected, we recorded the frequency and type of contractions in exposed ureters of anesthetized animals. The number of contractions was unusually higher in miR-143/145-defficient mice compared to wild-type controls. However peristaltic waves were replaced by incomplete, short and rapid contractions that failed to propagate in a proximal-distal direction. These alterations were already present in embryonic life. Microarray analysis uncovered candidate genes that regulate the contractility and propagation of the ureteric peristalsis.
We are exploring whether “rescue therapy” can be instituted by restoring the missing genomic miR145 to the ureter smooth muscle layer and thus return normal contractility and curing hydronephrosis medically (Figure 4). Simultaneously and in collaboration with the department of Pediatric Urology we are exploring the prevalence of this mutation in the children affected by hydronephrosis.
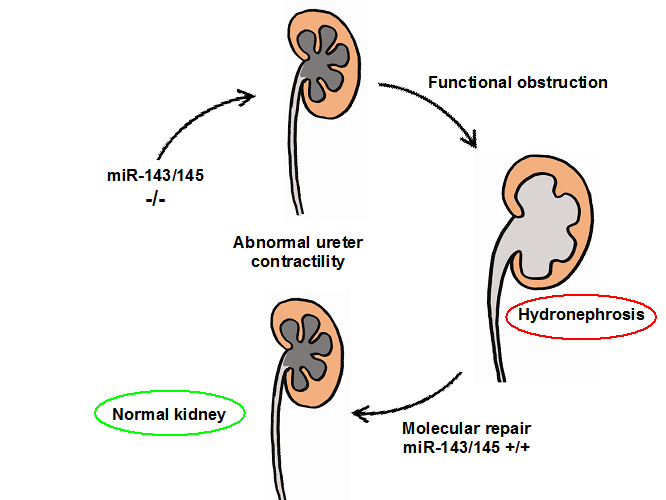
Figure 4
A. The BRenin cell, a progenitor for lymphoid leukemia.
Brian Belyea, MD
Maria Luisa Sequeira Lopez, MD
Ariel Gomez, MD
The Notch/RBP-J signaling pathway regulates multiple cell lineage decisions, including fate specification of renin precursor cells within the kidney. We have previously detected the presence of renin-expressing cells within the bone marrow and hypothesized that Notch/RBP-J may be similarly involved in lineage specification of these cells. During hematopoiesis, RBP-J dictates lymphocyte fate by promoting T cell development over B cell fate from common progenitors by repressing a network of B-lineage associated transcription factors. However, a causal role for Notch/RBP-J deficiency in B cell malignancies has not been previously demonstrated. In this study, we characterized the effect of RBP-J deficiency in cells from the renin lineage on the fate of hematopoietic precursor cells. To delete RBP-J in renin-lineage cells, we crossed our previously established Ren1dCre mice with “floxed” RBP-J mice. Mice with deletion of RBP-J in cells from the renin lineage developed signs and symptoms of leukemia including hepatosplenomegaly, lymphadenopathy, anemia, and leukocytosis. Evaluation demonstrated leukemic infiltration in multiple organs and progressive solid tumor development, ultimately resulting in early mortality at an average age of 9 months. Peripheral blood smears contained a predominance of lymphoblasts (Figure 5), and flow cytometry and lineage studies confirmed a cell autonomous B cell leukemia.
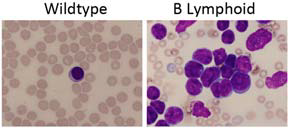 Figure 5
Figure 5
Characterization of the cell of origin in this model indicated that a small portion of B lymphocyte progenitors within the bone marrow arose from the renin lineage, we termed them the BRenin cells. Gene expression profiling performed on mRNA isolated from control and affected spleens demonstrated overexpression of genes involved in B cell commitment (IGLL1, Rag1, VpreB1) and repression of T cell related genes (TCRg-V1, CD7, CCR9). In addition, gene enrichment analysis showed overexpression of genes involved in cell cycle progression and proliferation. In conclusion, these studies demonstrate that the bone marrow contains a population of hematopoietic progenitors, which is derived from the renin lineage, and that RBP-J functions as a novel tumor suppressor within these cells. When RBP-J is silenced, there is de-repression of a B cell gene program leading to increased transcription of B-lineage associated genes. In addition, these cells develop a selective, proliferative advantage ultimately leading to B cell leukemia.
B. Epigenetic marks, cell phenotype and the leukemic state
Ellen S Pentz, Ph.D.
Ariel Gomez, MD
Experiments in our lab indicate that leukemic cell progenitors have altered chromatin conformation, which ultimately leads to the activation of oncogenes and /or deactivation of tumor suppressors. We performed a whole genome epigenetic landscape and uncovered a consistent pattern of H3K4me3 along the genome favoring genes whose transcripts are elevated in the leukemic cells while remaining silent or down-regulated in non-cancerous cells (Figure 6). Those genes are now the focus of potential screening for therapy using leukemic colony assays as shown below in Figure 7.
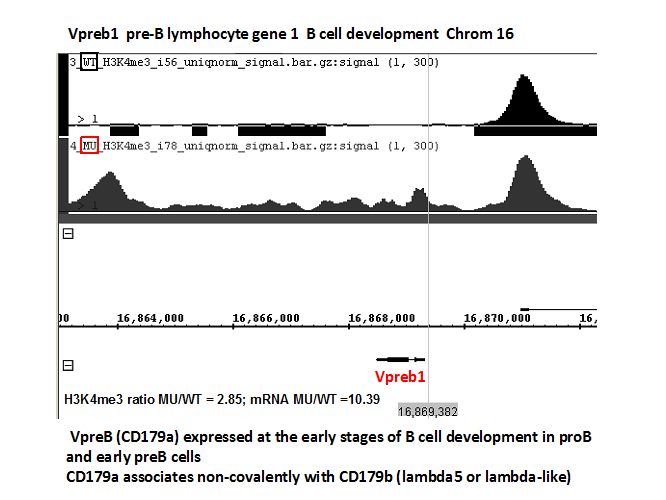 Figure 6
Figure 6
C. Identifying therapeutic compounds using colony assays from leukemic marrow.
Silvia Medrano, Ph.D.
Yan Hu, Graduate Student
Maria Luisa Sequeira Lopez, MD
Brian Belyea, MD
Ariel Gomez, MD
As we identify the crucial molecules that drive cells to become cancerous, we will develop and test therapeutic compounds on leukemic cells in culture. Bone marrow cells have the ability to form colonies as shown in Figures 7 and 8 below. In fact, leukemic cells have a growing advantage in that their colonies are larger and more numerous (Figure 7, L3, bottom right). As a proof of principle to examine whether our approach would be feasible, we treated leukemic and control colonies with vinblastine a well-known microtubule inhibitor that prevents cell replication. As shown on the top panel, vinblastine (+VB) completely inhibited colony growth of leukemic cells. We intend to use this strategy to screen a large number of chemical compounds, including microRNAs that may be supporting the cancer phenotype. Those compounds effective in vitro using GFP+ leukemic colonies will be also tested in vivo in mice with full-blown leukemia (Figures 8,9). This dual (in vitro-in vivo) strategy has enormous potential to identify not only the molecular pathogenesis of the disease but to specifically target those molecules that cause or maintain the disease as well as those that could serve as biomarkers of disease progression or regression.
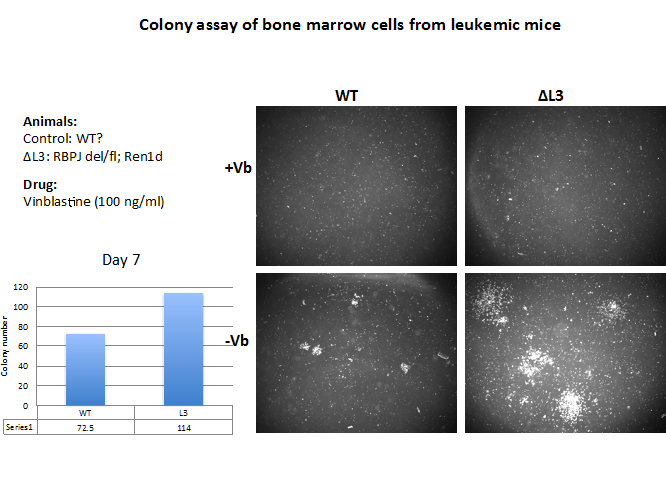 Figure 7
Figure 7
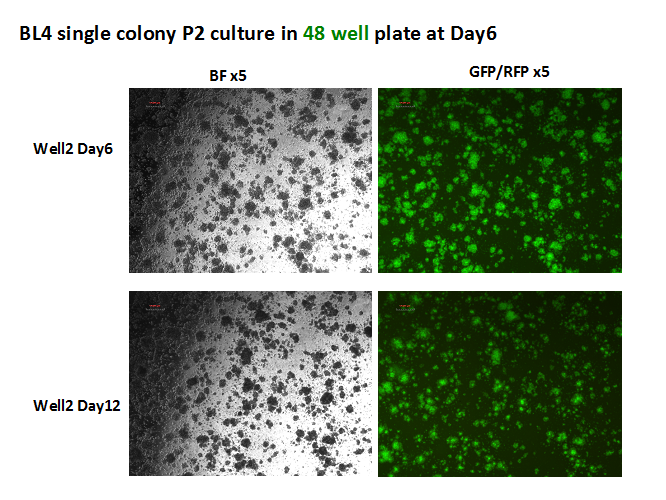 Figure 8: GFP+ colonies from the bone marrow of a mouse with pre B cell Leukemia. These colonies are being used to test which molecules and mechanisms sustain the disease phenotype and devise therapeutic compounds as shown below.
Figure 8: GFP+ colonies from the bone marrow of a mouse with pre B cell Leukemia. These colonies are being used to test which molecules and mechanisms sustain the disease phenotype and devise therapeutic compounds as shown below.
 Figure 9: GFP+ colonies in culture will be used to define the mechanism underlying this type of leukemia and to devised therapeutic compounds that can be tested in vivo using the leukemic mice we have generated thus far.
Figure 9: GFP+ colonies in culture will be used to define the mechanism underlying this type of leukemia and to devised therapeutic compounds that can be tested in vivo using the leukemic mice we have generated thus far.
Ellen. S Pentz, Ph.D.
Ariel Gomez, MD
We have shown that changes in the chromatin landscape in the renin locus determine the identity and function of the renin gene. In the model shown below (Figure 10), we show that increases in intracellular cAMP lead to acetylation of histone 4 around the cAMP responsive element in the renin enhancer that activated transcription of the renin gene and subsequent increase in renin release resulting in the control of body fluids and blood pressure homeostasis.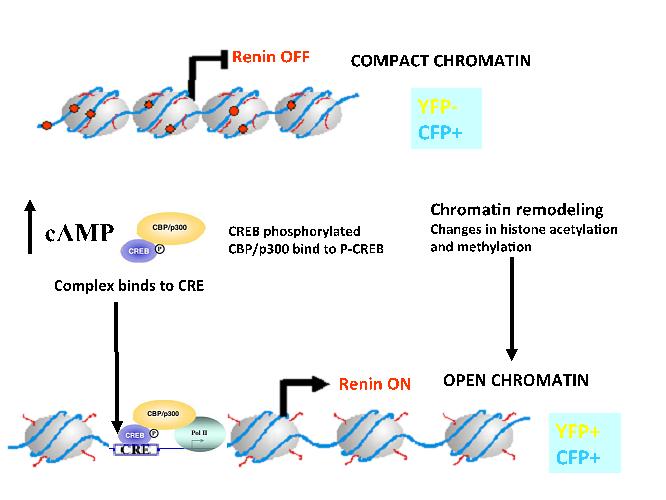
Figure 10